RFP Lab:
-Purpose:
-Make RFP from jelly fish in bacteria -Learn about steps of genetic engineering
-Materials + Procedure:
-Refer to Lab Note Book (Ex: 2a materials + procedure in Amgen lab manual part 2a)
-All Labs being stated on this page can be referenced to Lab Note Book
-Make RFP from jelly fish in bacteria -Learn about steps of genetic engineering
-Materials + Procedure:
-Refer to Lab Note Book (Ex: 2a materials + procedure in Amgen lab manual part 2a)
-All Labs being stated on this page can be referenced to Lab Note Book
Lab 2a:
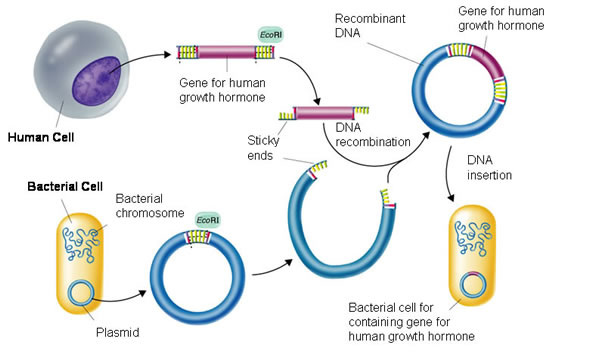
-Experimental Overview: Verification of plasmid by restriction digest; Cut plasmid (circular DNA) with BamHI + Hind III to cut out RFP-ara from bacterial plasmid.
-Results: My group (Rachel, Jacob) and I successfully cut out the RFP using the BamHI and Hind III and then reinserted it into the new recombinant plasmid. The RFP fit in the new recombinant plasmid because we had made a gap in the new recombinant plasmid using the same BamHI and Hind III that were used to cut the RFP out of its original plasmid. The RFP connected with the new plasmid because of the specificity of the sticky ends. Once the sticky ends on the new plasmid and RFP connected, the ligase repaired the phosphorus backbone of the DNA, securing the connection between the RFP and new recombinant plasmid.
-2a Questions:
~Stop and Think:
1.) Why does using two different enzymes to cut the plasmid prevent the plasmid from reforming a circle without the inserted gene?
A: Using two different enzymes creates different sticky ends on either side of this new gap in the plasmid. If the same enzyme had been used to cut both sides, both sides would be the same sticky ends and therefore stick together, closing the gap.
2.) Why are the enzymes placed in 37 degrees Celsius water?
A: Because that is body temperature and the enzymes will work best at body temperature.
~Before the Lab:
1.) If pARA-R is digested with BamHI and HindIII, what fragments are produced? Record the nucleotide sequence of the sticky ends and the length of each fragment (bp), and indicate the genes and other important sequences present on each fragment.
A: RFP fragment with pBAD and an Ara-C fragment with ori and Amp-R. The nucleotide sequence length of the RFP fragment is 807 BP and the Ara-C fragment is 4495 BP.
2.) In order to create a plasmid that can produce the red fluorescent protein in bacteria, what components are needed in the plasmid?
A: The RFP gene as well as Ara-C,which binds to the promoter region, are needed.
3.) Bacteria can be killed by an antibiotic unless they carry a plasmid that has the gene for resistance to that antibiotic. Biotechnologists call these genes selectable markers because only bacteria that carry the gene will survive an antibiotic. If the uptake of DNA by bacteria is inefficient, why is a selectable marker critical in cloning a gene in bacteria?
A: A selectable marker is important to cloning genes in bacteria because it determines which bacteria will continue to grow. Without the marker, the bacteria dies while the ones with it survive. When cloning a gene, you only want the bacteria that actually received the gene to pass on, and selectively marking them is a way to do that.
~During Lab:
1.) List in words or indicate in a drawing the important features of a plasmid vector that are required to clone a gene. Explain the purpose of each feature.
A: A plasmid vector is a plasmid that carries a certain trait. The important features include a selective marker, so that you can "select" only the successful transformation. Another feature is the ori, or origin of replication. The ori allows for replication of the plasmid when the bacterial cell divides. A promoter is another important feature that binds the DNA polymerase so that the selected gene can be expressed in the cell. Lastly, the restriction enzyme recognition site is needed. This is a gap in the plasmid where the desired gene may be inserted.
2.) What role do restriction enzymes have in nature?
A: Restriction enzymes are defense mechanisms. For example, if a virus inserts its DNA into a bacterium, the restriction enzymes will chop it up because it does not match the bacterium DNA.
3.) Using your understanding of evolution, why would bacteria retain a gene that gives them resistance to antibiotics? How is the existence of bacteria with antibiotic resistance affecting medicine today?
A: Bacteria would retain a gene that retains a resistance to antibiotics because only the ones the survive the antibiotics replicate. The medical fields constantly have to create new antibiotics because the old antibiotics have selected for the bacteria with resistance to the antibiotics.
4.) Bacteria, sea anemones, and humans seem, on the surface, to be very different organisms. Explain how a gene from humans or a sea anemone can be expressed in bacteria to make a product never before made in bacteria.
A: Central dogma, which is DNA to mRna to protein, is the reason human genes can be expressed in bacteria. The code of life is universal.
5.) Due to a mishap in the lab, bacteria carrying a plasmid with an ampicillinresistant gene and bacteria carrying a plasmid with a gene that provides resistance to another antibiotic (kanamycin) were accidentally mixed together. Design an experiment that will allow you to sort out the two kinds of bacteria.
A: Split the original petri dish into two petri dishes. Put the ampicillin in one and the kanamycin in the other, and it result in one petri dish that has killed off all the ampicillin sensitive cells, but kept the ampicillin resistant cells. Vise versa for the kanamycin sensitive cells in the other petri dish.
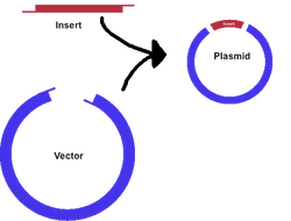
This is where we took the RFP (the insert) out of the vector and put it into the recombinant plasmid.
-Results: My group (Rachel, Jacob) and I successfully cut out the RFP using the BamHI and Hind III and then reinserted it into the new recombinant plasmid. The RFP fit in the new recombinant plasmid because we had made a gap in the new recombinant plasmid using the same BamHI and Hind III that were used to cut the RFP out of its original plasmid. The RFP connected with the new plasmid because of the specificity of the sticky ends. Once the sticky ends on the new plasmid and RFP connected, the ligase repaired the phosphorus backbone of the DNA, securing the connection between the RFP and new recombinant plasmid.
-2a Questions:
~Stop and Think:
1.) Why does using two different enzymes to cut the plasmid prevent the plasmid from reforming a circle without the inserted gene?
A: Using two different enzymes creates different sticky ends on either side of this new gap in the plasmid. If the same enzyme had been used to cut both sides, both sides would be the same sticky ends and therefore stick together, closing the gap.
2.) Why are the enzymes placed in 37 degrees Celsius water?
A: Because that is body temperature and the enzymes will work best at body temperature.
~Before the Lab:
1.) If pARA-R is digested with BamHI and HindIII, what fragments are produced? Record the nucleotide sequence of the sticky ends and the length of each fragment (bp), and indicate the genes and other important sequences present on each fragment.
A: RFP fragment with pBAD and an Ara-C fragment with ori and Amp-R. The nucleotide sequence length of the RFP fragment is 807 BP and the Ara-C fragment is 4495 BP.
2.) In order to create a plasmid that can produce the red fluorescent protein in bacteria, what components are needed in the plasmid?
A: The RFP gene as well as Ara-C,which binds to the promoter region, are needed.
3.) Bacteria can be killed by an antibiotic unless they carry a plasmid that has the gene for resistance to that antibiotic. Biotechnologists call these genes selectable markers because only bacteria that carry the gene will survive an antibiotic. If the uptake of DNA by bacteria is inefficient, why is a selectable marker critical in cloning a gene in bacteria?
A: A selectable marker is important to cloning genes in bacteria because it determines which bacteria will continue to grow. Without the marker, the bacteria dies while the ones with it survive. When cloning a gene, you only want the bacteria that actually received the gene to pass on, and selectively marking them is a way to do that.
~During Lab:
1.) List in words or indicate in a drawing the important features of a plasmid vector that are required to clone a gene. Explain the purpose of each feature.
A: A plasmid vector is a plasmid that carries a certain trait. The important features include a selective marker, so that you can "select" only the successful transformation. Another feature is the ori, or origin of replication. The ori allows for replication of the plasmid when the bacterial cell divides. A promoter is another important feature that binds the DNA polymerase so that the selected gene can be expressed in the cell. Lastly, the restriction enzyme recognition site is needed. This is a gap in the plasmid where the desired gene may be inserted.
2.) What role do restriction enzymes have in nature?
A: Restriction enzymes are defense mechanisms. For example, if a virus inserts its DNA into a bacterium, the restriction enzymes will chop it up because it does not match the bacterium DNA.
3.) Using your understanding of evolution, why would bacteria retain a gene that gives them resistance to antibiotics? How is the existence of bacteria with antibiotic resistance affecting medicine today?
A: Bacteria would retain a gene that retains a resistance to antibiotics because only the ones the survive the antibiotics replicate. The medical fields constantly have to create new antibiotics because the old antibiotics have selected for the bacteria with resistance to the antibiotics.
4.) Bacteria, sea anemones, and humans seem, on the surface, to be very different organisms. Explain how a gene from humans or a sea anemone can be expressed in bacteria to make a product never before made in bacteria.
A: Central dogma, which is DNA to mRna to protein, is the reason human genes can be expressed in bacteria. The code of life is universal.
5.) Due to a mishap in the lab, bacteria carrying a plasmid with an ampicillinresistant gene and bacteria carrying a plasmid with a gene that provides resistance to another antibiotic (kanamycin) were accidentally mixed together. Design an experiment that will allow you to sort out the two kinds of bacteria.
A: Split the original petri dish into two petri dishes. Put the ampicillin in one and the kanamycin in the other, and it result in one petri dish that has killed off all the ampicillin sensitive cells, but kept the ampicillin resistant cells. Vise versa for the kanamycin sensitive cells in the other petri dish.
This is where we took the RFP (the insert) out of the vector and put it into the recombinant plasmid.
Lab 4a:
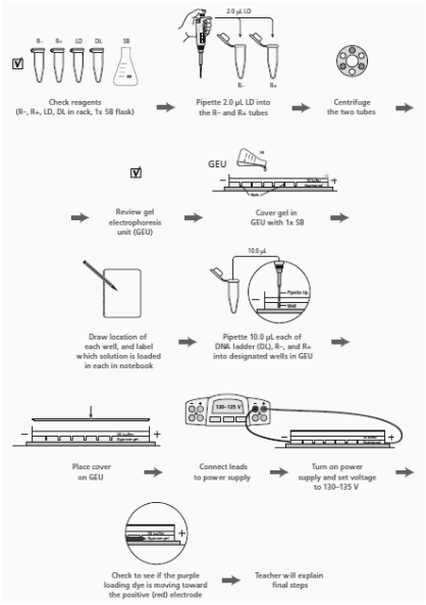
-Experimental Overview: Verification of plasmid digest by electrophoresis to ensure the plasmid was cut correctly before. We ran the plasmid in a gel alongside a DNA ladder. A DNA ladder has different-sized molecules already programmed into it, so after being run, it provides a scale that you can compare whatever you are running on the gel to. We already knew the size of the RFP gene and the plasmid, so when we ran it, it should have matched that known size, we could tell if it did by comparing what we ran to the DNA ladder, and seeing if it matched the correct (known) size.
-Results: The DNA ladder was unsuccessful, but the plasmid with the RFP successfully split into 2 bands and the plasmid without RFP into one, verifying we correctly had done step 2a.
-4a questions:
~Review:
1.) Why do DNA restriction fragments and plasmids separate when analyzed by gel electrophoresis?
A: Because they have different charges and sizes.
2.) Why is it important to identify and verify a recombinant plasmid?
A: Because you want to make sure you have successfully inserted the gene into the new plasmid.
~Stop and Think:
1.) The DNA is not visible as it moves through the gel. The loading dye contains the three dyes that you separated in Laboratory 1. Why is it useful to use the loading dye in this lab?
A: Because we need to be able to clearly see our DNA on our gels and dye makes that DNA more distinct and visible to the human eye.
2.) The DNA samples and the DNA ladder are not visible on the gel.How might the DNA be made visible once the gel electrophoresis is complete?
A: You could shine UV light through the DNA and the dye will illuminate.
~Before the Lab:
1.) The pARA-R plasmid you digested in Laboratory 2A was replicated in a bacterial cell. What configurations (supercoiled, nicked circle, and multimer) might the plasmid have before digestion?
A: The plasmid could be all three: supercoiled, nicked circle, and multimer.
2.) You need to catalog all the products you might see, including the different plasmid configurations. Review your work in Laboratory 2A. What products might you expect to see in the R– and R+ tubes? Create a table that shows all the possible fragments and plasmids by tube. Include the length (bp size) of each possible fragment or plasmid, and arrange the products found in each microfuge tube by size, from smallest to largest. Include any possible plasmid configurations, and arrange them first by size and next by speed through the gel, from fastest to slowest.
-Results: The DNA ladder was unsuccessful, but the plasmid with the RFP successfully split into 2 bands and the plasmid without RFP into one, verifying we correctly had done step 2a.
-4a questions:
~Review:
1.) Why do DNA restriction fragments and plasmids separate when analyzed by gel electrophoresis?
A: Because they have different charges and sizes.
2.) Why is it important to identify and verify a recombinant plasmid?
A: Because you want to make sure you have successfully inserted the gene into the new plasmid.
~Stop and Think:
1.) The DNA is not visible as it moves through the gel. The loading dye contains the three dyes that you separated in Laboratory 1. Why is it useful to use the loading dye in this lab?
A: Because we need to be able to clearly see our DNA on our gels and dye makes that DNA more distinct and visible to the human eye.
2.) The DNA samples and the DNA ladder are not visible on the gel.How might the DNA be made visible once the gel electrophoresis is complete?
A: You could shine UV light through the DNA and the dye will illuminate.
~Before the Lab:
1.) The pARA-R plasmid you digested in Laboratory 2A was replicated in a bacterial cell. What configurations (supercoiled, nicked circle, and multimer) might the plasmid have before digestion?
A: The plasmid could be all three: supercoiled, nicked circle, and multimer.
2.) You need to catalog all the products you might see, including the different plasmid configurations. Review your work in Laboratory 2A. What products might you expect to see in the R– and R+ tubes? Create a table that shows all the possible fragments and plasmids by tube. Include the length (bp size) of each possible fragment or plasmid, and arrange the products found in each microfuge tube by size, from smallest to largest. Include any possible plasmid configurations, and arrange them first by size and next by speed through the gel, from fastest to slowest.
|
3.) Read through the Methods section on pages 64 through 66 and briefly outline the steps, using words and a flowchart. ---->
~During the Lab: 1.) Why is it important to verify that you have the correct recombinant plasmid? A: So that the gene you are going for is in the plasmid and then can actually be expressed, as you were intending. If you do not verify that you have the correct plasmid, all your next steps will fail. 2.) How did your actual gel results compare to your gel predictions? A: Our R+ and R- columns came out in the correct spots (2 bands for the recombinant and 1 band for the plasmid without the RFP), yet were a little difficult to see. 3.) Do you see any bands that are not expected? What could explain the origin of these unexpected bands? A: There were a few unexpected bands. These could have originated when more of the plasmid was digested then expected. 4.) Does the gel show that you are using the correct recombinant plasmid? Describe the evidence you used to make this assessment. A: The gel does show that the R+ lane has two bands, however our DNA ladder was not present so it was impossible to see if they were the correct size. 5.) In the R– lane, do you see evidence of multiple configurations of plasmids? Explain your answer. A: In the R- lane I see evidence of multiple configurations of plasmids because the plasmids had not yet been modified to a nicked circle. 6.) In the R+ lane, do you see evidence of complete digestion? Explain your answer. A: Yes in the R+ lane the fact that there are two different bands shows complete digestion because that proves that the RFP and the plasmid separated. 7.) In which lane would you expect to find the rfp gene and the ampR gene in the gel photograph? Are you able to locate these two genes? Explain your answer. A: I would expect to find the RFP gene in the R+ lane because that is where the plasmid had RFP. Same goes for the AmpR. In the R+ lane, we expect to see a band around 807 bp as well as a band at around 4,495 bp. I could not locate them because the dye did not work all that well. 8.) Compare the lanes that have linear fragments with the lanes that have plasmids. Is there a difference in the shape of the bands between these two DNA forms? A: With linear DNA there is only one band, however with the plasmid it was able to be digested into multiple parts. The bands for the linear fragments have trailing edges unlike the non trailing edges of the lanes with plasmids. |
|

Our result, but the DNA ladder wasn't present due to an error in our experiment
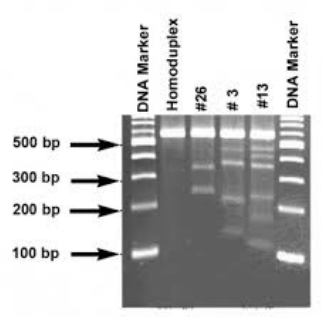
Example of DNA Ladder with successful results
Lab 5a:
-Experimental Overview: Transformation of bacteria with recombinant plasmid. We now inserted the recombinant plasmid into the bacteria. This was done by inserting calcium chloride into the bacteria and providing a heat shock. This made a hole for the recombinant plasmid to inert into the bacteria. We then used a selective marker (Amp-R, resistance to Ampicillin) to select only the successfully inserted bacteria.
-Results: The calcium chloride and heat shock did the trick for some bacteria. Then, the selective marking was successful.
-5a Questions:
~Stop and Think:
1.) How is the P+ bacteria culture treated differently from the P–bacteria culture? (A culture is an isolated population of cells.) What is the purpose of the P– bacteria culture?
A: The P+ is treated differently in that is must be kept separate from the P-. The P- is there t prove that the ampicillin will kill the P- bacteria but not the P+ bacteria.
2.) Why do the cells need time to recover after the heat shock?
A: So they do not overheat and die.
3.) Why are the cells incubated at 37°C?
A: Because that is the human body temperature at which cells grow best.
4.) You will use aseptic technique in this lab. Why is this important?
A: It helps us to not cross contaminate the P+ and P- bacteria.
~Before the Lab:
1.) Ampicillin is an antibiotic that kills bacterial cells by disrupting the formation of cell walls. However, the pARA-R plasmid has the ampicillin resistance gene, which produces a protein that breaks down ampicillin. What is the purpose of growing bacteria that have been transformed in the presence of ampicillin?
A: The purpose of growing these bacteria is it separates those with the resistance from those without it.
2.) What will happen when bacterial cells that contain the pARA-R plasmid are not given arabinose?
Without arabinose the RFP gene will not be expressed so there will be no red glow.
3.) In the lab, you will add samples of the control group P– and the treatment group P+ to plates that contain various combinations of Luria Broth (LB), ampicillin, and the sugar arabinose. Predict the growth on each dish.
A: I predict that Plate I (LB) will have non-glowing growth on both sides. I predict that Plate II (LB/Amp) will have non-glowing growth on the P+ side. For Plate III (LB/Amp/Ara), I predict a glowing red colony on the whole plate.
~During the Lab:
1.) Look at the results of your transformation. Do your actual results match your predicted results? If not, what differences do you see, and what are some explanations for these differences? (reference picture underneath with clear plates)
A: Our results matched our predictions in almost every way. However,, the colony on the LB/Amp/Ara plate was not glowing red as it should have. This was not our fault, as the company we got our supplies from provided us with faulty material.
2.) How many red colonies were present on your LB/amp/ara plate?
A: There were no red colonies present on our plate.
3.) Why did the red colonies only appear on the LB/Amp/Ara plate and not the LB/Amp plate?
A: The red colonies would have only appeared on the plate with araganose because araganose is the promoter which allows the red protein to be expressed.
4.) Recombinant plasmids are engineered so that they replicate independently from the replication of the chromosomes. Why is it important to have multiple copies of a recombinant plasmid within a cell?
A: The more plasmids in a cell means the more production and the more likely chance of successfully passing on those genes in a plasmid.
5.) How is the information encoded in the RFP gene expressed as a trait? Be sure to use what you have previously learned about gene expression and the relationship between DNA, RNA, Protein, and traits.
A: Because of central dogma (DNA makes RNA which makes Protein). The promoter(ara) binds the DNA of the RFP gene to the bacterias DNA. This makes a new code, which makes a new RNA, and that RNA is the RNA that makes RFP protein.
-Results: The calcium chloride and heat shock did the trick for some bacteria. Then, the selective marking was successful.
-5a Questions:
~Stop and Think:
1.) How is the P+ bacteria culture treated differently from the P–bacteria culture? (A culture is an isolated population of cells.) What is the purpose of the P– bacteria culture?
A: The P+ is treated differently in that is must be kept separate from the P-. The P- is there t prove that the ampicillin will kill the P- bacteria but not the P+ bacteria.
2.) Why do the cells need time to recover after the heat shock?
A: So they do not overheat and die.
3.) Why are the cells incubated at 37°C?
A: Because that is the human body temperature at which cells grow best.
4.) You will use aseptic technique in this lab. Why is this important?
A: It helps us to not cross contaminate the P+ and P- bacteria.
~Before the Lab:
1.) Ampicillin is an antibiotic that kills bacterial cells by disrupting the formation of cell walls. However, the pARA-R plasmid has the ampicillin resistance gene, which produces a protein that breaks down ampicillin. What is the purpose of growing bacteria that have been transformed in the presence of ampicillin?
A: The purpose of growing these bacteria is it separates those with the resistance from those without it.
2.) What will happen when bacterial cells that contain the pARA-R plasmid are not given arabinose?
Without arabinose the RFP gene will not be expressed so there will be no red glow.
3.) In the lab, you will add samples of the control group P– and the treatment group P+ to plates that contain various combinations of Luria Broth (LB), ampicillin, and the sugar arabinose. Predict the growth on each dish.
A: I predict that Plate I (LB) will have non-glowing growth on both sides. I predict that Plate II (LB/Amp) will have non-glowing growth on the P+ side. For Plate III (LB/Amp/Ara), I predict a glowing red colony on the whole plate.
~During the Lab:
1.) Look at the results of your transformation. Do your actual results match your predicted results? If not, what differences do you see, and what are some explanations for these differences? (reference picture underneath with clear plates)
A: Our results matched our predictions in almost every way. However,, the colony on the LB/Amp/Ara plate was not glowing red as it should have. This was not our fault, as the company we got our supplies from provided us with faulty material.
2.) How many red colonies were present on your LB/amp/ara plate?
A: There were no red colonies present on our plate.
3.) Why did the red colonies only appear on the LB/Amp/Ara plate and not the LB/Amp plate?
A: The red colonies would have only appeared on the plate with araganose because araganose is the promoter which allows the red protein to be expressed.
4.) Recombinant plasmids are engineered so that they replicate independently from the replication of the chromosomes. Why is it important to have multiple copies of a recombinant plasmid within a cell?
A: The more plasmids in a cell means the more production and the more likely chance of successfully passing on those genes in a plasmid.
5.) How is the information encoded in the RFP gene expressed as a trait? Be sure to use what you have previously learned about gene expression and the relationship between DNA, RNA, Protein, and traits.
A: Because of central dogma (DNA makes RNA which makes Protein). The promoter(ara) binds the DNA of the RFP gene to the bacterias DNA. This makes a new code, which makes a new RNA, and that RNA is the RNA that makes RFP protein.
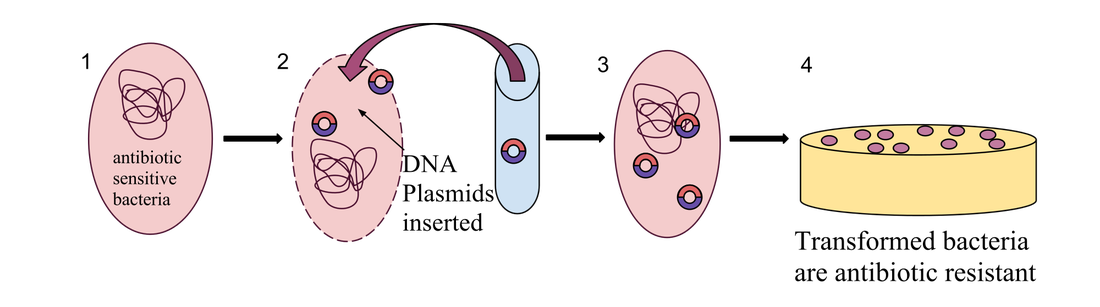
Recombinant plasmid inserted into plate to create Bacteria
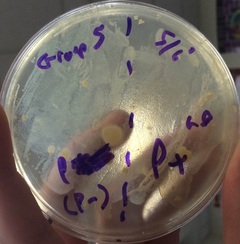
Plate 1
|
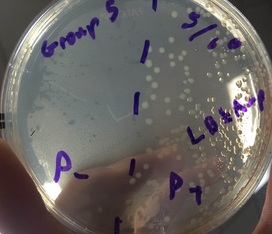
Plate 2
|
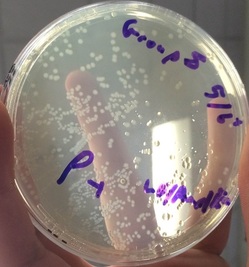
Plate 3
|
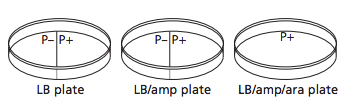
-Plates Explained: In the first picture we have inserted a single recombinant plasmid into a single bacteria, however, in real life, you really are not going to be that skilled. So instead you take a decent amount of bacteria and use the enzymes and heat shock hopping that holes will form in the bacteria for the recombinant plasmids to enter. However, this does not happen in most of the bacteria. So only a few bacteria now contain the recombinant plasmid. Now you want just the bacteria with the recombinant plasmid in them, but how do you get just those ones. Remember how on that recombinant plasmid there is a ampicillin resistant gene. So if you pour some ampicillin into the tray of all the bacteria, the only survivors will be the ones with the recombinant plasmid. The trays on the right show ow only certain parts with the plasmid grew. Oh and the LB is a growth nutrient for the bacteria and the ara is a gene (RFP) promotor.
LAB 6a:
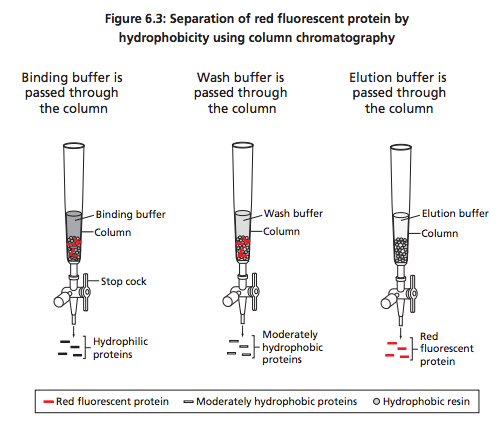
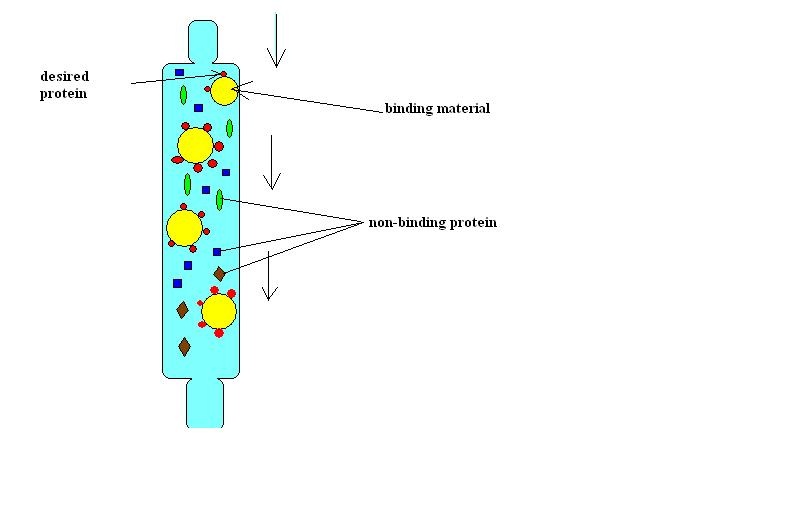
-Experimental Overview: Purification of RFP using chromatography. Now that the bacteria have grown the RFP for us through incubation(growth with supplied nutrients), we need to separate the RFP. We used a chromatography column with hydrophilic beads, this collected the RFP in the resin bed. We then eluted the RFP and separated it from the beads using an enzyme.
-Results: We were able to successfully remove the RFP proteins from the other parts of the lysed bacteria cells.
-6a Questions:
~Stop and Think:
1.) How can you determine where the red fluorescent protein is in each separation step??
A: The RFP will be red and will centrifuge to the bottom.
2.) What color is the supernatant? The pellet? What are the contents of each?
A: The supernatant is a clear liquid (usually composed of solutions) found on top of a recently microfuged solid. The pellet is this solid and is often made of proteins(RFP).
~Before the Lab:
1.) How can solutions of different salt concentrations, which will unfold proteins to varying degrees, be used to help purify red fluorescent protein using column chromatography?
A: Proteins are unfolded in the highly salt concentrated buffer. The proteins that don't flow out the column (because they are unfolded and to big now) are refolded when lower salt concentrations are added, thus then they leave the column later.
~During the Lab:
1.Why is a protein’s conformation important for carrying out its function?
A: A protein's conformation is important because it determines the promoter regions of the given protein. These promoter regions are what give each protein their different functions.
2.) What properties of the amino acids in a protein relate to protein folding?
A: The sequence of an amino acid determines how it will fold into a protein.
3.) Does the eluate containing your red fluorescent protein appear less bright or brighter than it did in the cell lysate following centrifugation? If there is a noticeable difference in the intensity of the red color, what might account for that?
A: The RFP-containg elute is brighter than the cell lysate. This is because the elute contains the most RFP, the cell lysate contained all the cell proteins and cell parts along with the RFP.
4.) What characteristic of red fluorescent protein is used as the basis for separation by column chromatography?
A: The red fluorescent protein sticks to the resin column when unfolded (more hydrophobic amino acids).
5.) How might the column chromatography procedure be adjusted or modified to increase the purity of the red fluorescent protein sample?
A: If we repeated the process with more wash buffers and were more careful about collection, we could increase the red fluorescent protein purity. Also, the size and shape of the column can be modified to maximize good results.
-Results: We were able to successfully remove the RFP proteins from the other parts of the lysed bacteria cells.
-6a Questions:
~Stop and Think:
1.) How can you determine where the red fluorescent protein is in each separation step??
A: The RFP will be red and will centrifuge to the bottom.
2.) What color is the supernatant? The pellet? What are the contents of each?
A: The supernatant is a clear liquid (usually composed of solutions) found on top of a recently microfuged solid. The pellet is this solid and is often made of proteins(RFP).
~Before the Lab:
1.) How can solutions of different salt concentrations, which will unfold proteins to varying degrees, be used to help purify red fluorescent protein using column chromatography?
A: Proteins are unfolded in the highly salt concentrated buffer. The proteins that don't flow out the column (because they are unfolded and to big now) are refolded when lower salt concentrations are added, thus then they leave the column later.
~During the Lab:
1.Why is a protein’s conformation important for carrying out its function?
A: A protein's conformation is important because it determines the promoter regions of the given protein. These promoter regions are what give each protein their different functions.
2.) What properties of the amino acids in a protein relate to protein folding?
A: The sequence of an amino acid determines how it will fold into a protein.
3.) Does the eluate containing your red fluorescent protein appear less bright or brighter than it did in the cell lysate following centrifugation? If there is a noticeable difference in the intensity of the red color, what might account for that?
A: The RFP-containg elute is brighter than the cell lysate. This is because the elute contains the most RFP, the cell lysate contained all the cell proteins and cell parts along with the RFP.
4.) What characteristic of red fluorescent protein is used as the basis for separation by column chromatography?
A: The red fluorescent protein sticks to the resin column when unfolded (more hydrophobic amino acids).
5.) How might the column chromatography procedure be adjusted or modified to increase the purity of the red fluorescent protein sample?
A: If we repeated the process with more wash buffers and were more careful about collection, we could increase the red fluorescent protein purity. Also, the size and shape of the column can be modified to maximize good results.
|
|
Example of RFP Lab
FINAL STEP:
-Experimental Overview: Now that we have collected our RFP protein from the bacteria, there is one last final part. We need to once again run a gel, only this time with the RFP protein. We run the protein next to a Protein ladder to check if it the right size. And if there is only single bands it means the protein is pure because there is only one type of protein.
-Results: After working on this lab for months (since the beginning of the year), the RFP was pure and the right size and the lab was complete!
-Results: After working on this lab for months (since the beginning of the year), the RFP was pure and the right size and the lab was complete!

-Analysis & Conclusion:
In this lab, we tried to make a red glowing bacteria colony. We tried to do this be inserting the gene for a red fluorescent protein (RFP) into the DNA of a bacteria. However, due to bad materials from our lab suppliers, our proteins did not glow. Through verification and selective marking, we knew that we had successfully inserted the gene into the bacteria, but once again, our supplies were a little faulty. After we had inserted the gene we wanted into the bacterium through recombinant plasmids, we nurtured the cells to grow our protein. This works because of central dogma. Then, we separated the RFP proteins from the rest of the bacterium through lyse (breaking open the cell with enzymes) and column chromatography.
-Reflection:
This lab was extremely hard for me. I was really confused throughout the lab experiments, but I don't think it's anyone else's fault but my own. I should have taken more notes than I thought I did and spend more time understanding the answer to the questions then just writing them out. I understand what the lab was manly for, making a red bacteria colony, but don't quite understand the bases of it.
I think I should have asked more questions. I am always scared to ask questions because I don't tend to grasp something I don't understand unless someone spells it out for me. Plus when people let me ask, I ask LOTS of questions when I don't understand it. I like making sure it's right every 2 seconds. I could be told something hundreds of times how to do it and I will still question if I am doing it correctly. I also don't like it when people act like it's simple, when really it isn't for me.
My group was tuff. Not because they were off task a lot (which we never really did anyway), but because Rachel and I were both confused how to answer the questions and take the notes. Jacob was having difficulties understanding everything too. I don't blame them for my ignorance though. They were both trying, but Jacob understood everything a lot better in the end after spending more time on trying to understand it. I look at this as a lesson. I shouldn't do nothing and expect things to change, that's LITERALLY the definition of insanity. I should have spent more time on understand something, then sitting around stressed out instead of doing something productive.
Couldn't have done it without my group members. In the next lab I do, I promise to focus on the lab, not focus on getting it over with.
In this lab, we tried to make a red glowing bacteria colony. We tried to do this be inserting the gene for a red fluorescent protein (RFP) into the DNA of a bacteria. However, due to bad materials from our lab suppliers, our proteins did not glow. Through verification and selective marking, we knew that we had successfully inserted the gene into the bacteria, but once again, our supplies were a little faulty. After we had inserted the gene we wanted into the bacterium through recombinant plasmids, we nurtured the cells to grow our protein. This works because of central dogma. Then, we separated the RFP proteins from the rest of the bacterium through lyse (breaking open the cell with enzymes) and column chromatography.
-Reflection:
This lab was extremely hard for me. I was really confused throughout the lab experiments, but I don't think it's anyone else's fault but my own. I should have taken more notes than I thought I did and spend more time understanding the answer to the questions then just writing them out. I understand what the lab was manly for, making a red bacteria colony, but don't quite understand the bases of it.
I think I should have asked more questions. I am always scared to ask questions because I don't tend to grasp something I don't understand unless someone spells it out for me. Plus when people let me ask, I ask LOTS of questions when I don't understand it. I like making sure it's right every 2 seconds. I could be told something hundreds of times how to do it and I will still question if I am doing it correctly. I also don't like it when people act like it's simple, when really it isn't for me.
My group was tuff. Not because they were off task a lot (which we never really did anyway), but because Rachel and I were both confused how to answer the questions and take the notes. Jacob was having difficulties understanding everything too. I don't blame them for my ignorance though. They were both trying, but Jacob understood everything a lot better in the end after spending more time on trying to understand it. I look at this as a lesson. I shouldn't do nothing and expect things to change, that's LITERALLY the definition of insanity. I should have spent more time on understand something, then sitting around stressed out instead of doing something productive.
Couldn't have done it without my group members. In the next lab I do, I promise to focus on the lab, not focus on getting it over with.
